
| Idiopathische pulmonale fibrose | ||||
|---|---|---|---|---|
| Coderingen | ||||
| ICD-10 | J84.1 | |||
| ICD-9 | 516.3 | |||
| OMIM | 178500 | |||
| DiseasesDB | 4815 | |||
| MedlinePlus | 000069 | |||
| eMedicine | radio/873 | |||
| MeSH | D011658 | |||
| ||||
Idiopathische pulmonale fibrose (IPF), oftewel idiopathische longfibrose is een chronische en uiteindelijk dodelijke ziekte die zich kenmerkt door een progressieve achteruitgang van de longfunctie. Het begrip longfibrose betekent bindweefselvorming in het longweefsel en is de oorzaak van verergerende dyspneu (kortademigheid). Fibrose heeft doorgaans een slechte prognose. Het begrip 'idiopathisch' wordt gebruikt omdat de oorzaak van longfibrose nog onbekend is.
IPF komt het meeste voor bij volwassenen boven de 50 jaar, en met name bij mensen die roken of gerookt hebben. De ziekte komt meer voor bij mannen dan bij vrouwen.
IPF behoort tot een grote groep van meer dan 200 longziekten die interstitiële longziekten (ILD) worden genoemd, met de betrokkenheid van het long interstitium als gemeenschappelijk kenmerk. Het interstitium, het weefsel tussen de longblaasjes in de long, is waar de schade bij ILD's primair optreedt. In veel gevallen tasten deze ziekten echter niet alleen het interstitium aan, maar ook de longblaasjes, perifere luchtwegen en bloedvaten. Longweefsel van mensen met IPF vertoont een kenmerkend histopathologisch patroon dat 'usual interstitial pneumonia' (UIP) wordt genoemd.
In 2011 zijn nieuwe richtlijnen voor de diagnose en behandeling van IPF gepubliceerd. Voor de diagnose van IPF moeten andere bekende oorzaken van ILD worden uitgesloten en moet er sprake zijn van een kenmerkend radiologisch patroon dat zichtbaar wordt gemaakt door middel van computertomografie met hoge resolutie (HRCT). Met de juiste klinische faciliteiten is het mogelijk om IPF uitsluitend met HRCT te diagnosticeren, waardoor het niet nodig is om via chirurgie een longbiopsie uit te voeren.
Classificatie
IPF behoort tot de interstitiële longziekten, waarbij de betrokkenheid van het long interstitium, een gemeenschappelijk kenmerk is. IPF is één specifiek ziektebeeld binnen de groep van idiopathische interstitiële pneumoniën (IIP), die ook wel diffuse parenchymatische longziekten (DPLD) genoemd worden.
IPF onderscheidt zich van de overige IIP's doordat het de enige ziekte is binnen de groep, waarvan de oorzaak onbekend is. Andere vormen van IIP zijn onder meer niet-specifieke interstitiële pneumonie (NSIP), desquamatieve interstitiële pneumonie (DIP) en acute interstitiële pneumonie (AIP).
Voorbeelden van ILD met een bekende oorzaak zijn onder meer allergische alveolitis, langerhanscelhistiocytose in de longen, asbestose en collageen-vasculaire ziekten. In veel gevallen tasten deze ziekten echter niet alleen het interstitium aan, maar ook de longblaasjes, perifere luchtwegen en bloedvaten.
Epidemiologie
IPF is weliswaar zeldzaam, maar toch de meest voorkomende vorm van IIP. In Nederland lijden naar schatting 2.500-3.000 mensen aan IPF, gebaseerd op een gemiddelde prevalentie van 16,7 per 100.000 inwoners. IPF komt vaker voor bij mannen dan bij vrouwen en de diagnose wordt doorgaans gesteld bij mensen van 50 jaar en ouder.
In de (toenmalige) 27 landen van de Europese Unie komt uit een reeks verschillende bronnen een incidentie naar voren van 4,6-7,4 per 100.000 inwoners. In Nederland betekent dit dat er gebaseerd op een gemiddelde incidentie van 6,0 per 100.000 jaarlijks ongeveer 1000 nieuwe IPF-patiënten bij komen.
Oorzaken van/risicofactoren voor IPF
Zoals de naam al aangeeft, is IPF een idiopathische aandoening. Dat wil zeggen dat de oorzaak onbekend is. Er zijn overigens wel bepaalde omgevingsfactoren bekend die mogelijk een relatie hebben met het ontstaan ervan, zoals blootstelling aan bepaalde stoffen. Roken is de bekendste en breedst geaccepteerde risicofactor voor IPF en verhoogt het risico op IPF met ongeveer een factor twee. [10] Ook blootstelling aan andere omgevings- en beroepsgerelateerde stoffen, zoals metaalstof, houtstof, silicium, steenstof en blootstelling aan stoffen in bijvoorbeeld de agrarische sector kunnen het risico op IPF doen toenemen. Er zijn ook aanwijzingen dat (virus)infecties mogelijk verband houden met IPF en andere fibrotische longziekten.
Etiologie en pathofysiologie
Ondanks uitvoerig onderzoek is de oorzaak van IPF nog steeds onbekend. De fibrose die bij IPF optreedt, wordt in verband gebracht met sigarettenrook, omgevingsfactoren (bijv. beroepsgerelateerde blootstelling aan gassen, rook, chemicaliën of stof), andere medische aandoeningen als oesofageale reflux, of genetische aanleg. Geen van deze factoren geldt echter voor alle mensen met IPF en daarom vormen ze geen volledig sluitende verklaring voor de ziekte.
Van IPF wordt vermoed dat het een gevolg is van een verkeerd verlopend (wond)genezingsproces, waarbij sprake is van een afwijkende reactie op een mogelijk herhaalde trigger. Dit leidt tot overmatige afzetting van collageen (fibrose) in het longinterstitium met samengaand met enige mate van ontsteking.
Verondersteld wordt dat bij IPF de oorspronkelijke (herhaalde) trigger de longcellen, oftewel alveolaire epitheelcellen (AEC's, pneumocyten) aantast. Met deze cellen is het grootste gedeelte van het alveolaire oppervlak bekleed. Wanneer AEC's van type I beschadigen of verloren gaan, zo denkt men, vindt proliferatie van AEC's van type II plaats om de blootliggende basale membranen te bedekken. Bij normaal herstel sterven de hyperplastische AEC's van type II af, waarna de resterende cellen zich verspreiden en een differentiatieproces ondergaan om te veranderen in AEC’s type I. Onder pathologische omstandigheden en bij aanwezigheid van transformerende groeifactor bèta (TGFβ), hopen fibroblasten zich op in deze beschadigde delen, waarbij ze veranderen in myofibroblasten die collageen en andere proteïnen afgeven. In het verleden werd gedacht dat een ontsteking de aanzet vormde voor littekenvorming in longweefsel. De meest recente bevindingen wijzen er echter op dat de ontwikkeling van fibroblastische foci voorafgaat aan het ontstaan van concentraties ontstekingscellen en de daaropvolgende afzetting van collageen. Deze hypothese wordt indirect ondersteund door de klinische verschijnselen van IPF, zoals een ongemerkt ontstaan, progressie gedurende meerdere jaren, relatief zeldzame acute exacerbaties en ongevoeligheid voor immunosuppressiva. Voor een aantal therapieën gericht op activering van fibroblasten of de synthese van extracellulaire matrix zijn de eerste onderzoeksfasen gaande of wordt ontwikkeling overwogen.
Familiale IPF betreft minder dan 5% van alle IPF-patiënten en is uit klinisch en histologisch oogpunt niet te onderscheiden van sporadische IPF. Genetische associaties zijn onder meer surfactantproteïnen A1, A2, C (SFTPA1, SFTPA2B) en mucine (MUC5B). Mutaties van humane telomerasegenen worden ook in verband gebracht met familiale longfibrose en in sommige gevallen met sporadische IPF (TERT, TERC).
Diagnose
Een vroegtijdige diagnose van IPF is een voorwaarde voor snellere behandeling en, in potentie, verbetering van het klinische resultaat op de lange termijn bij deze progressieve en uiteindelijk dodelijke ziekte. Wanneer IPF wordt vermoed, kan de diagnose een lastig proces zijn, maar het is gebleken dat de nauwkeurigheid van de IPF-diagnostiek verbetert als een multidisciplinaire aanpak wordt gehanteerd waaraan wordt bijgedragen door een longarts, een radioloog en een pathologisch expert op het gebied van interstitiële longziekten.
Een multidisciplinaire consensusverklaring over idiopathisch interstitiële pneumonieën, in 2002 uitgebracht door de American Thoracic Society (ATS) en de European Respiratory Society (ERS) omvat een voorstel voor specifieke major en minor criteria voor het diagnosticeren van IPF. In 2011 zijn echter nieuwe, vereenvoudigde en geactualiseerde criteria uitgebracht voor de diagnose en begeleiding van IPF door de ATS en ERS, in samenwerking met de Japanese Respiratory Society (JRS) en de Latin American Thoracic Association (ALAT). Op dit moment gelden voor een IPF-diagnose de volgende criteria:
- Het uitsluiten van bekende vormen van ILD, zoals beelden die passen bij blootstelling aan een specifieke stof, bijvoorbeeld de ‘asbestlong’, bindweefselstoornissen of blootstelling aan/toxiciteit van geneesmiddelen en straling;
- De aanwezigheid van een kenmerkend radiologisch UIP-patroon op HRCT.
Met de juiste klinische faciliteiten is het mogelijk om IPF uitsluitend met HRCT te diagnosticeren, waardoor het niet nodig is om via chirurgie een longbiopsie uit te voeren.
Het herkennen van IPF in de klinische praktijk kan lastig zijn, omdat de symptomen vaak lijken op die van algemener voorkomende ziekten, zoals astma, chronische obstructieve longziekte (COPD) en hartfalen. Het voornaamste probleem waar artsen voor staan, is de vraag of de medische achtergrond, symptomen (of verschijnselen), radiologie en longfunctieonderzoek als geheel aansluiten bij de diagnose van IPF, dan wel of de bevindingen verband houden met een ander proces. Al geruime tijd wordt erkend dat gevallen van ILD door blootstelling aan asbest, geneesmiddelen (zoals chemotherapeutica of nitrofurantoïne), reumatoïde artritis en sclerodermie/systemische sclerose moeilijk van IPF te onderscheiden zijn. Andere differentiële diagnostische afwegingen zijn onder meer interstitiële longziekten, waaronder gevallen die verband houden met mixed connective tissue disease, gevorderde sarcoïdose, chronische allergische alveolitis, langerhanscelhistiocytose in de longen en bestralingspneumonitis.
Klinische verschijnselen

Bij veel patiënten is al geruime tijd sprake van symptomen voordat de diagnose wordt gesteld. Tot de meest voorkomende klinische verschijnselen van IPF behoren:
- leeftijd 50 jaar of ouder;
- droge, niet-productieve hoest;
- voortschrijdende inspanningsdyspneu (benauwdheid bij inspanning);
- typische droge, inspiratoire bibasale crepitaties bij auscultatie, namelijk een krakerig geluid lijkend op klittenband dat langzaam wordt losgetrokken; dit wordt ook 'velcrocrackles' genoemd, naar het Engelse woord voor klittenband: velcro;
- trommelstokvingers en/of -tenen (zie afbeelding);
- afwijkende resultaten bij longfunctieonderzoek, met bewijs voor restrictieve longaandoening en belemmerde gaswisseling.
De meeste van deze verschijnselen houden verband met chronisch zuurstofgebrek in het bloed en zijn niet specifiek voor IPF. Ze komen ook voor bij tal van andere long- of hartaandoeningen. IPF dient te worden overwogen bij alle patiënten met onverklaarde chronische inspanningsdyspneu, hoesten, bibasale crepitaties bij inademen, of trommelstokvingers.
Beoordeling van de kenmerkende crepitaties ('klittenbandgeluid') bij longauscultatie is een praktische manier om de diagnose IPF eerder te kunnen stellen. Wanneer artsen dit typische geluid al eens gehoord hebben, is het eenvoudig te herkennen en het is kenmerkend voor IPF.
|
|
|
| IPF longgeluid (download·info) |
Als bij een oudere patiënt gedurende de hele inademing crepitaties te horen zijn en deze na meerdere diepe ademhalingen aanhouden, en bij vervolgonderzoek enige weken later nog evengoed hoorbaar zijn, kan dat op IPF wijzen en is er aanleiding om een HRCT-scan van de borstkas te overwegen (vanwege de hogere gevoeligheid dan een thoraxfoto). Omdat crepitaties op zich niet specifiek zijn voor IPF, dienen ze aanleiding te zijn voor een grondig diagnostisch proces.
Indien deze symptomen worden waargenomen, moeten de juiste nadere onderzoeken worden verricht om IPF te kunnen diagnosticeren.
Radiologie
Thoraxfoto's zijn nuttig bij de vaste nacontroles bij IPF-patiënten. Zij zijn helaas niet geschikt voor diagnostiek, maar kunnen wel verminderde longvolumes zichtbaar maken, eventueel, in een gevorderd stadium, met prominente reticulaire interstitiële patronen nabij de longbases.

Bij IPF is de radiologische evaluatie door middel van HRCT een essentieel onderdeel van het diagnostische traject. HRCT vindt plaats met een conventionele CT-scanner zonder injectie van contrastmiddelen. De te evalueren doorsnedes zijn altijd zeer dun (1–2 mm). Bij IPF zijn op HRCT-beelden van de thorax de kenmerkende fibrotische veranderingen in beide longen te zien, met name in de longbases en de periferie. Volgens de richtlijnen in de gezamenlijke ATS/ERS/JRS/ALAT 2011, is HRCT een essentieel onderdeel van het diagnostische traject bij IPF, waarmee UIP kan worden vastgesteld op grond van de aanwezigheid van:
- Reticulaire verdichtingen, vaak samengaand met tractiebronchiëctasieën;
- Honingraatpatroon (‘honeycombing’), oftewel een cluster van luchthoudende cysteuze structuren met een duidelijke wand, doorgaans vergelijkbaar in diameter (3–10 mm) maar soms ook groter (tot 2,5 cm). Meestal subpleuraal gelegen en gevormd in ten minste twee lijnen. In het algemeen is één lijn van cysten niet voldoende om van een honingraatpatroon te kunnen spreken;
- Matglasverdichtingen komen vaak voor, maar zijn minder aanwezig dan de reticulatie;
- Kenmerkend is de basale en perifere locatie van de afwijkingen;

Histologie
Op grond van de herziene richtlijnen van 2011 geldt dat, indien HRCT geen UIP-patroon uitwijst, een operatieve longbiopsie noodzakelijk is om tot een betrouwbare diagnose te kunnen komen.
Histologische monsters voor de diagnose van IPF moeten op ten minste drie verschillende plaatsen worden afgenomen, en dienen groot genoeg te zijn om de patholoog in staat te stellen de onderliggende opbouw van het longweefsel te beoordelen. Kleine biopten, die bijvoorbeeld worden verkregen door middel van een transbronchiale longbiopsie (uitgevoerd tijdens een bronchoscopie), zijn doorgaans niet toereikend voor dit doeleinde. Ook niet omdat IPF zich vooral in de periferie van de long voorkomt en bij een transbronchiaal biopt gemist kan worden. Daarom is het meestal noodzakelijk om grotere biopten af te nemen via een thoracotomie of thoracoscopie.
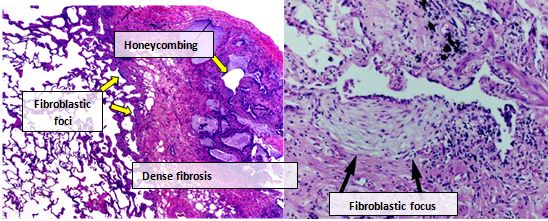
Longweefsel van mensen met IPF vertoont doorgaans een kenmerkend histopathologisch UIP patroon en UIP is dan ook de pathologische equivalent van IPF. Hoewel een pathologische diagnose van UIP vaak overeenkomt met een klinische diagnose van IPF, kan een histopathologisch UIP-patroon ook voorkomen bij andere ziekten (bijvoorbeeld reumatische aandoeningen). Er zijn vier belangrijke kenmerken van UIP: interstitiële fibrose met heterogene distributie, dat wil zeggen ongelijkmatige aantasting van het longweefsel door fibrose interstitiële littekenvorming, honingraatstructuren, en fibroblastische foci. Fibroblastische foci zijn clusters van myofibroblasten en littekenweefsel, en vormen in combinatie met honingraatpatroon de voornaamste pathologische bevindingen die een diagnose van UIP mogelijk maken.
Broncho-alveolaire lavage
Broncho-alveolaire lavage (BAL) is een diagnostische procedure, die gebruikt kan worden bij ILD. Voor de evaluatie van patiënten met IPF kan naar het inzicht van de behandelend arts een BAL-cytologie analyse worden overwogen op basis van de beschikbaarheid en ervaring in de desbetreffende instelling. BAL kan IPF niet aantonen, maar wel alternatieve specifieke diagnoses aan het licht brengen: maligniteit, infecties, eosinofiele pneumonie, langerhanscelhistiocytose, of alveolaire proteïnose. Bij de evaluatie van patiënten met vermoedelijke IPF ligt de belangrijkste toepassing van BAL in de uitsluiting van andere diagnoses. Prominente lymfocytose (>40%) doet bijvoorbeeld vermoeden dat er kans is op extrinsieke allergische alveolitis/hypersensitivity pneumonitis.
Longfunctieonderzoek
Spirometrie toont in de meeste gevallen een vermindering van de vitale capaciteit (VC) en een evenredige vermindering van geforceerde vitale capaciteit (FVC) Meting van statische longvolumes via lichaamsplethysmografie of andere technieken laat in de meeste gevallen verminderde longvolumes (restrictie) zien. Deze meting laat zien hoeveel moeite het kost om de fibrotische longen met lucht te vullen. De diffusiecapaciteit voor koolstofmonoxide (DLCO) is bij IPF altijd verminderd en kan bij lichte vormen of in vroege stadia van de ziekte het enige afwijkende aspect zijn. Dit wordt ook zichtbaar in de vatbaarheid van IPF-patiënten voor zuurstofdesaturatie bij inspanning, die ook kan worden onderzocht met de 6-minutenwandeltest (6MWT) .
Begrippen als ‘mild’, ‘matig’ en ‘ernstig’ worden soms gebruikt om de ziekte in stadia te verdelen en zijn doorgaans gebaseerd op metingen van longfunctietests in rusttoestand. Er bestaat echter nog geen duidelijke consensus over de IPF-stadiëring bij patiënten en de criteria en waarden die daarvoor het best kunnen worden gebruikt. Milde tot matige IPF wordt soms gedefinieerd als voldoende aan de volgende functionele criteria:
- Geforceerde vitale capaciteit (FVC) ≥50%;
- DLCO ≥35%;
- 6MWT-afstand ≥150 meter.
Prognose

Het klinisch beloop van IPF kan onvoorspelbaar zijn. De progressie van IPF wordt in verband gebracht met een geschatte mediane overlevingsduur van twee tot vijf jaar na diagnose. Bij IPF loopt de 5-jaarsoverleving uiteen van 20–40%. Daarmee ligt dit mortaliteitscijfer hoger dan dat van een aantal maligne aandoeningen, waaronder dikkedarmkanker, multipel myeloom en blaaskanker.
Onlangs is een multidimensionaal index- en stadiëringssysteem voorgesteld om de mortaliteit bij IPF te voorspellen. Deze index, GAP genaamd, is gebaseerd op geslacht [G], leeftijd [A] en twee longfysiologische variabelen [P] (FVC and DLCO) die in de klinische praktijk in de meeste gevallen worden gemeten om de mortaliteit bij IPF te voorspellen. Het hoogste GAP-stadium (stadium III) wordt in verband gebracht met een mortaliteitsrisico van 39% na 1.
Behandeling
De behandeling van IPF beoogt in essentie vermindering van de symptomen, het stilleggen van de progressie, preventie van acute exacerbaties en verlenging van de overlevingsduur. Preventieve zorg (bijv. griepvaccinaties) en behandeling op symptoombasis moeten bij elke patiënt in een vroeg stadium worden gestart.
Farmacologische interventies
- Pirfenidon
In het verleden zijn diverse behandelingen voor IPF onderzocht, waaronder interferon gamma-1β,bosentan, ambrisentan, en anticoagulantia , maar die werden ineffectief bevonden. Veel van deze eerdere onderzoeken waren gebaseerd op de hypothese dat IPF een ontstekingsziekte is.
Pirfenidon is een klein molecuul dat in experimentele modellen van fibrose een combinatie van ontstekingsremmende, antioxidatieve en antifibrotische effecten vertoont. Pirfenidon is in Europa, India, Japan en China goedgekeurd voor de behandeling van patiënten met lichte tot matige IPF. Het is op de markt onder de handelsnamen Esbriet (in Europa), Pirfenex (in India), Pirespa (in Japan) en Etuary (in China).
Pirfenidon is in de Europese Unie goedgekeurd op basis van de resultaten van drie gerandomiseerde, dubbelblinde, placebo-gecontroleerde fase III-studies, waarvan er één in Japan en de andere twee in Europa en de VS zijn uitgevoerd (CAPACITY-studies).
Een evaluatie van de Cochrane Library (van The Cochrane Collaboration, een internationale organisatie die zich inzet voor Evidence-Based Medicine (EBM)), gebaseerd op vier onderzoeken met 1155 patiënten waarin pirfenidon is vergeleken met een placebo, heeft een significant lager (30%) risico van ziekteprogressie aangetoond bij patiënten die met pirfenidon werden behandeld. Een vertraging van de FVC-afname, die ook na een jaar nog significant was, kon echter in slechts een van de twee CAPACITY-onderzoeken worden aangetoond. Op basis van deze gemengde resultaten heeft de Amerikaanse Food and Drug Administration (FDA) verzocht om een derde fase III-onderzoek, dat de titel ASCEND (NCT01366209) draagt en is uitgevoerd in de VS en een aantal kleinere landen.
De resultaten van ASCEND zijn in mei 2014 in The New England Journal of Medicine gepubliceerd. Het primaire eindpunt van de studie was het verschil in verlies van FVC tussen de met pirfenidon behandelde groep en de met placebo behandelde groep. Het verlies in FVC was 45,1% minder in de met pirfenidon behandelde groep. Dit komt overeen met een verminderd absoluut verlies van 193 ml. Daarnaast is vergeleken hoeveel procent van de patiënten in beide groepen meer dan 10% FVC verloren of overleden. Dit is een belangrijke parameter, omdat een verlies van meer dan 10% FVC een sterk verhoogd risico geeft op overlijden. Het percentage patiënten dat meer dan 10% FVC verloor of overleed was in de pirfenidon groep 47,9% lager. Ook de 6 minutenloopafstand (6MWD) en progressievrije overleving waren beide significant beter in de pirfenidon-groep. In de dyspneuscore (mate van benauwdheid) en de sterftecijfers was het verschil tussen beide groepen niet significant. In een vooraf vastgestelde analyse waarin de gegevens van de ASCEND en de CAPACITY-studies gezamenlijk werden geanalyseerd, werd aangetoond dat pirfenidon de mortaliteit significant deed afnemen: de afname van sterfte door welke oorzaak dan ook was 48%, de afname van sterfte aan idiopathische longfibrose was 68%.
- N-acetylcysteïne en drievoudige therapie
N-acetylcysteïne (NAC) speelt een rol in de productie van glutathion, een antioxidant. Verondersteld wordt dat de oxidant-antioxicantbalans in het longweefsel van IPF-patiënten verstoord is en dat behandeling met hoge doses NAC kan zorgen voor herstel van de deze balans. In een klinische onderzoek met 180 patiënten (IFIGENIA) is gebleken dat toevoegen van NAC aan de combinatie prednison en azathioprine (drievoudige therapie), de afname van de VC en DLCO verminderde gedurende een controleperiode van 12 maanden in vergelijking met de groep patiënten die geen NAC toegevoegd kregen aan hun therapie met prednison en azathioprine.
In Amerika is vervolgens door de National Institutes of Health een gerandomiseerd onderzoek (PANTHER-IPF) uitgevoerd bij patiënten met IPF ter beoordeling van bovengenoemde drievoudige therapie in vergelijking met alleen NAC of helemaal geen therapie (placebo).. Uit dat onderzoek bleek dat de combinatie van prednison, azathioprine en NAC het risico van ziekenhuisopname en overlijden verhoogde. De NIH maakte in 2012 bekend dat de onderzoeksarm voor drievoudige therapie van het PANTHER-IPF-onderzoek voortijdig werd afgebroken. Het onderzoek in de patiëntengroepen die alleen NAC of alleen een placebo kregen is wel voortgezet en de resultaten zijn in 2014 gepubliceerd. De conclusie van het onderzoek was dat “vergeleken met een placebo, N-acetylcysteïne geen significante voordelen bood ten aanzien van het behoud van de FVC bij patiënten met idiopathische longfibrose met lichte tot matige verslechtering van de longfunctie”. Gezien deze beperkingen wordt momenteel in de meest recente IPF-richtlijnen voor het merendeel van de patiënten met IPF noch drievoudige therapie noch NAC-monotherapie aanbevolen.
- Toekomstige behandelingsopties
Voor een aantal beoogde therapieën zijn de eerste onderzoeksfasen gaande of wordt ontwikkeling overwogen. Deze moleculen zijn gericht tegen diverse groeifactoren en cytokinen waarvan bekend is dat ze een rol spelen bij de proliferatie, activering, differentiatie of ondoelmatige overleving van fibroblasten.
Behandelingen die momenteel worden geëvalueerd in verder gevorderde onderzoeken naar IPF zijn onder meer:
- Nintedanib (BIBF 1120) is onderzocht in het kader van twee fase III-onderzoeken.
- Een fase II-onderzoek naar STX-100 loopt op dit moment.
Meer informatie is te vinden op ClinicalTrials.gov, een register en resultatenbestand met gegevens over publiek en privaat ondersteunde klinische onderzoeken met menselijke deelnemers die wereldwijd worden/zijn uitgevoerd
Niet-farmacologische interventies
- Longtransplantatie
Longtransplantatie kan geschikt zijn voor patiënten die fysiek in aanmerking komen om een zware transplantatieoperatie te ondergaan. Aangetoond is dat longtransplantatie bij IPF-patiënten het overlijdensrisico met 75% verlaagt ten opzichte van patiënten die op de wachtlijst blijven.
Hoewel er geen consensus over bestaat, wijzen de meest recente gegevens er wel op dat bilaterale longtransplantatie superieur is aan enkelvoudige longtransplantatie bij patiënten met IPF. De 5-jaarsoverleving na longtransplantatie bij IPF wordt geschat op 50 tot 56%.
- Permanente zuurstoftherapie
In de IPF-richtlijnen van 2011 werd zuurstoftherapie, of aanvullende zuurstof voor thuisgebruik, een sterke aanbeveling voor patiënten met klinisch significante hypoxemie in rusttoestand. Hoewel van zuurstoftherapie niet is gebleken dat de overleving van IPF-patiënten erdoor verbetert, wijzen bepaalde gegevens wel op een verbetering van de inspanningscapaciteit.
- Longrevalidatie
Vermoeidheid en verlies van spiermassa zijn veel voorkomende en invaliderende problemen waar IPF-patiënten mee kampen. Longrevalidatie kan de uiterlijke symptomen van IPF verlichten en het functioneren verbeteren door het stabiliseren en/of ongedaan maken van deze extrapulmonale ziekteverschijnselen. Het aantal gepubliceerde onderzoeken naar de rol van longrevalidatie bij idiopathische longfibrose is gering, maar de meeste ervan hebben aangetoond dat er sprake is van significante kortetermijnverbeteringen van de functionele inspanningstolerantie, kwaliteit van leven en inspanningsdyspneu. Vaak toegepaste revalidatieprogramma's zijn onder meer fysieke training, voedingsaanpassingen, bezigheidstherapie, educatie en psychosociale hulp. In de late fase van de ziekte houden IPF patiënten vaak op met de fysieke activiteiten vanwege de toenemende dyspneu. Zo mogelijk dient dit te worden afgeraden.
Palliatieve zorg
Palliatieve zorg is gericht op verlichting van symptomen en verbetering van het comfort van patiënten, en niet op behandeling van de ziekte. Behandeling van verergerende symptomen met chronische toediening van morfinomimetica voor ernstige dyspneu en hoesten kan daar deel van uitmaken. Daarnaast kan zuurstoftherapie nuttig zijn voor de verlichting van dyspneu bij patiënten met hypoxemie.
Palliatieve zorg omvat tevens verlichting van fysiek en emotioneel lijden en psychosociale hulp voor patiënten en verzorgenden. Naarmate de ziekte vordert, kunnen patiënten angst, spanning en depressie ervaren, en in die gevallen moet psychologische ondersteuning worden overwogen. In een recent onderzoek onder poliklinisch behandelde patiënten met ILD's, waaronder IPF, droegen de depressiescore, functionele status (bepaald met wandeltest) en tevens de longfunctie allemaal bij aan de ernst van de dyspneu.
In bepaalde gevallen van uitgesproken ernstige dyspneu zou morfine kunnen worden overwogen. Dat kan dyspneu, angst en hoesten verlichten zonder significante afname van de zuurstofsaturatie.
Begeleiding en follow-up
Bij IPF wordt vaak een verkeerde diagnose gesteld, ten minste totdat fysiologische en/of beeldvormingsgegevens wijzen op de aanwezigheid van een ILD, waardoor gepaste zorg pas laat op gang komt. Aangezien IPF een ziekte is met een mediane overlevingsduur van drie jaar na diagnose, moet vroegtijdige doorverwijzing naar een instelling met specifieke expertise worden overwogen voor elke patiënt met een vermoedelijke of aangetoonde ILD. Gezien de complexe differentiële diagnostiek is het voor een juiste diagnose uitermate belangrijk dat er multidisciplinair overleg plaatsvindt tussen longartsen, radiologen en pathologen die ervaring hebben met ILD-diagnoses.
Nadat de diagnose IPF is gesteld en de juiste behandeling is gekozen op grond van symptomen en het stadium van de ziekte, dient een nauwkeurig vervolgtraject te worden ingesteld. Vanwege het sterk variabele beloop van de ziekte en de hogere incidentie van complicaties als longkanker (tot 25% van de patiënten, zo is voor IPF bekend) is standaard een drie- tot zesmaandelijks onderzoek verplicht, waar spirometrie (lichaamsplethysmografie), diffusiecapaciteitstest, thoraxfoto's, 6MWT, dyspneubeoordeling, kwaliteit van leven en zuurstofbehoefte deel van moeten uitmaken.
De toenemende kennis van complicaties en vaak voorkomende bijkomende aandoeningen bij IPF vergen daarnaast een standaard evaluatie van comorbiditeiten, waarvan de meeste de weerslag vormen van gelijktijdige ouderdomsaandoeningen en medicijngebruik in de vorm van interacties en bijwerkingen.
Acute exacerbaties
Onder acute exacerbaties van IPF (AE-IPF) wordt elke onverklaarde verergering of het ontstaan van dyspneu binnen 30 dagen verstaan, waarbij nieuwe radiologisch waarneembare (op HRCT beelden) infiltraten vaak te zien zijn tegen een achtergrond die overeenstemt met een UIP patroon. De jaarlijkse incidentie van AE-IPF ligt tussen de 10 en 15% van alle patiënten. De prognose van AE-IPF is slecht en de mortaliteit varieert tussen 78% en 96%. Andere oorzaken van AE-IPF, zoals longembolie, congestief hartfalen, pneumothorax of infectie, moeten worden uitgesloten. Longinfecties moeten worden uitgesloten aan de hand van endotracheaal aspiraat of BAL.
Veel patiënten bij wie een acute verergering optreedt, hebben een IC-behandeling nodig, met name wanneer respiratoire insufficiëntie verband houdt met hemodynamische instabiliteit, significante comorbiditeiten of ernstige hypoxemie. De mortaliteit tijdens ziekenhuisopnames is echter hoog. Mechanische beademing moet alleen worden toegepast als de langetermijnprognose en zo mogelijk de wensen van de patiënt zorgvuldig zijn afgewogen. De huidige richtlijnen raden het echter af om mechanische beademing toe te passen als de respiratoire insufficiëntie secundair is aan de IPF.
Bij dieren
IPF is een erkende aandoening bij een aantal honden- en kattenrassen en is het beste beschreven bij de West Highland White Terrier. Dieren met deze aandoening hebben veel klinische verschijnselen gemeen met hun menselijke equivalenten, waaronder progressieve inspanningsintolerantie, verhoogde ademfrequentie en uiteindelijk ademnood.
De prognose is over het algemeen slecht.
Externe links
Bronnen, noten en/of referenties
|